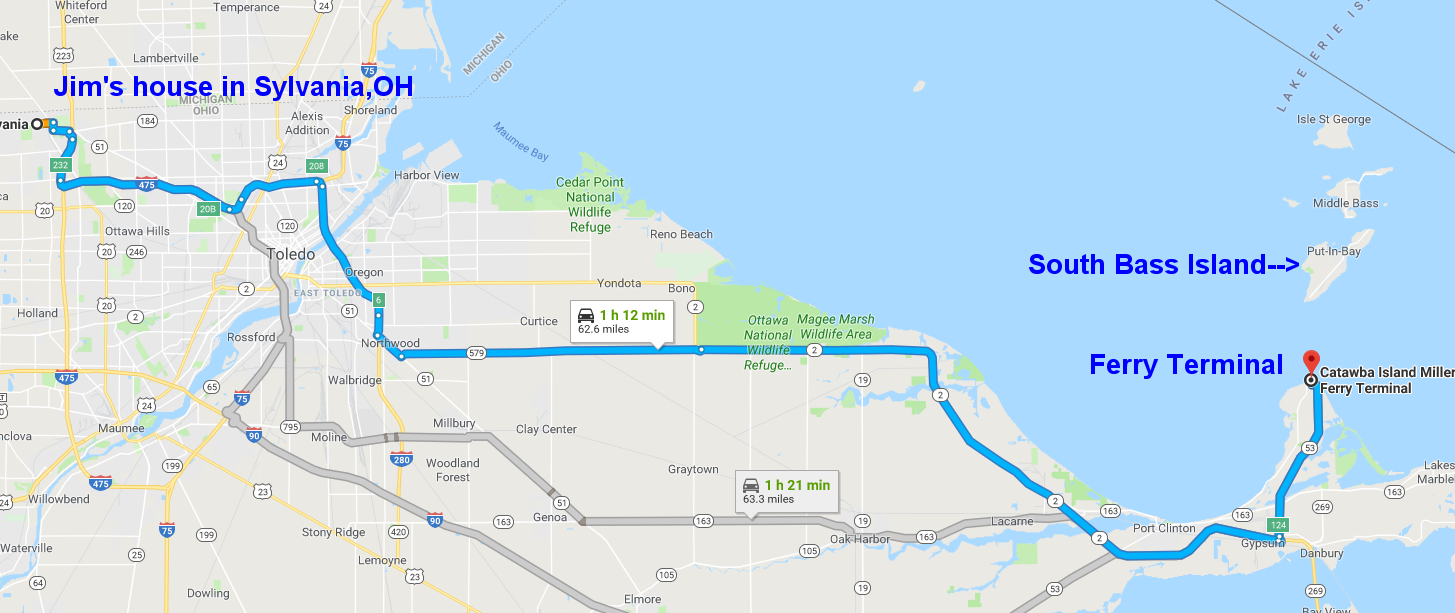
I originally wrote this blog a day or so after the Supreme Court decision overturning Roe leaked. I decided against posting it until I had time to reflect more. I concluded I would wait to release it until the actual decision was official. Here are my thoughts.
I’ve wanted to write this blog post for many years and I’ve hesitated to do so because I’m certain that many of my friends whose opinions, beliefs, and values I deeply respect will likely disagree with what I’m about to say. I need to get some things off my chest on the issue of abortion..
One of the reasons I’ve hesitated to express my views on the topic is that there is a large group of people who hold the position that as a man, I don’t get an opinion on this issue. I acknowledge that part of the problem is that men have been imposing their views on the subject for centuries. I recognize that men do not have as much of a deeply vested interest in the issue as women. But as a person who considers themselves to have a strong moral character and as a person of faith, I don’t think I should be prohibited from holding opinions on the topic nor prohibited from expressing them. So while I don’t want to be seen as another man who’s trying to butt into something that’s none of his business, I’m simply expressing my thoughts on the topic. I’m not trying to impose my views on anyone.
In general, I don’t want to disparage anyone for their beliefs on the issue either. On the other hand, I am willing to point out what I believe are instances of hypocrisy and disingenuous beliefs by many people.
It’s not my intent to stereotype either side. I’m sure many pro-choice people will say, “Maybe lots of pro-choice people are that way but not me.” Similarly, many pro-life people can legitimately say, “Maybe that’s a lot of pro-life people but it’s not me.” As divided as the two camps are, I want to clearly acknowledge that there are differences of opinion and strategy within both groups. I’m not saying that either camp is homogenous in their beliefs. I’m talking in general, broad terms. If the shoe doesn’t fit, don’t wear it. In the end, these are just my opinions and my observations.
What I Believe
I do not believe that human life begins at conception.
Even though this is contrary to the official teaching of the Catholic Church and even though I deeply respect the Church’s views on the topic, I suppose I would have to say that is a matter of faith and it’s not a particular faith that I hold. I will later make some philosophical arguments why one might hold that human life begins at conception even though it’s not a position I hold myself. I would do so to point out the fact that I do understand the reasoning behind the view. I simply do not believe that that reasoning is sufficient to change my opinion.
My mom used to say, “As Mason said to Dixon…’ You’ve got to draw the line somewhere.’” For me, I always believed that the Supreme Court’s decision in both Roe and Casey that draws the line at viability is a reasoned argument.
During his initial presidential campaign, Barack Obama when asked, “When does life begin?” replied something to the effect of, “That’s above my pay scale.” When push comes to shove, I like his answer. But rather than avoiding the question completely. I agree with the original Supreme Court decisions on viability.
Apart from the question of when human life actually begins, I deeply disapprove of abortion on philosophical grounds in most cases. I do not believe however that aborting a pre-viable fetus is the murder of a human being. I don’t mean to trivialize this but if pushed to describe what I think about it, I would classify it as, “a really bad idea.”
There are lots of things in this world that are really bad ideas that are not in any way illegal. There are things that I would strongly advise people not to do. Things that I would not engage in myself. But these “really bad ideas” do not rise to the level where I think they ought to be regulated or that choices to do them should be imposed upon others. This has nothing to do with women’s rights or men making a decision for women. There are lots of really bad ideas that I don’t support but I don’t condemn people for doing them.
One of the problems with the abortion debate is that it is often framed as a binary issue. One absolutely must be either pro-choice or pro-life. The debate is framed in such a way that there cannot be any middle ground. That is unfair. This is a complicated issue. And I find myself sitting squarely on the fence. I think that abortion is a decision that women ought to make for themselves. And it is my belief that the best choice is not to abort.
So, one of the things that people probably will not like about this entire essay is that I’m not taking a stand firmly and absolutely on either side. The only concession I’ve ever heard in the countless hours of public discourse on the topic came recently from Sen. Amy Klobuchar (D) Minn. When asked by a journalist if she thought that being pro-choice was a litmus test for future Democratic candidates, she replied that she knowledged there were people who were personally opposed to abortion but were not willing to impose that belief on other people. In effect, she was talking about me. It was the first time in decades of debate that I’ve heard a public official acknowledge the acceptability of someone who did not have an absolute all-or-nothing approach to the topic.
Ignoring the Core Issue
The thing that troubles me the most about the abortion debate is the way that neither side seems to acknowledge the central issue. They misrepresent each other’s views. The very names of the two factions in the debate frame the discussion in such a way that avoids the central issue.
The central issue should only be, “When does human life begin?”
When one describes themselves as “pro-life” in opposition to the other camp, it misrepresents the other side as being opposed to the sanctity of human life which is not at all the case. I don’t know anyone on the pro-choice side of the issue who says, “Indeed human life begins at conception but women ought to be free to murder an innocent child for whatever reason they want.” No one who is pro-choice believes that abortion is murder. No one who is pro-choice disrespects the sanctity of human life. We know this because one of the major arguments of the pro-choice position is that allowing easy access to safe abortion will save women’s lives. So declaring someone as not being “pro-life” is a misrepresentation of the pro-choice position.
Both sides are legitimately pro-life but those who are opposed to abortion fail to acknowledge that. Furthermore, I don’t think I’ve ever heard someone who is pro-choice stand up and say, “Yes I am pro-life but I disagree on when life begins.” They argue in favor of protecting the lives of women. But they won’t describe themselves as “pro-life” even though they are. Furthermore, I’m pretty sure that most people who are on the liberal pro-choice side of the argument also oppose the death penalty which is a further indication that they respect human life. Sadly, that’s a statement we cannot make as broadly among conservative pro-life advocates. The Catholic Church has a doctrine called, “The consistent ethic of life” and they are as vehemently opposed to the death penalty as they are opposed to abortion. Would that this be true among more alleged pro-life people. Conservative opposition to common sense gun control, universal healthcare, and other social justice issues also call into the question the appropriateness of describing themselves as “pro-life”
Similarly, one would hope that those who are opposed to abortion, do believe in personal freedom and the ability to live one’s life as they wish and to make personal choices so long as it does not harm other people. Sadly, many people who oppose abortion also seek to impose their views in other areas such as homosexuality, the use of non-abortive birth control, and other personal choice issues. When at its core, if you don’t conflate issues, there is nothing about believing that human life begins at conception that necessarily makes you anti-choice.
The inability of either side to see the common ground inflames the passionate demonization of each side. I firmly believe that there could be more reasoned debate and deeper understanding and even healing if both sides would acknowledge that they are both inherently pro-life and pro-choice.
Again, it’s not about choice. It’s not about respecting life. It’s about defining when life begins. So much of the debate disregards what the debate is really about at its core and that is deeply disappointing to a rational person such as me who tries to respect the morality and humanity of everyone. Both sides of the debate are guilty of this issue.
Demonizing Pro-Choice
The pro-life side of the debate often does emphasize their belief that human life begins at conception. And you may think I’m crazy for saying this, but I don’t think they say it enough. Whenever asked a question about the issue, they ought to say, “Because we are firmly committed to the truth that human life begins at conception, we believe that [fill in the blank].”
When they argue against pro-choice, they tend to demonize the position portraying them as the murders of innocent children.Yet they never acknowledge that the pro-choice side sincerely, completely, and totally does not believe that abortion is murder. I would prefer that when describing the other side, pro-life people would say, “We acknowledge that people in the pro-choice movement sincerely do not believe that abortion is murder.” They can then go on to talk about how misguided that view is in their opinion. While it might come across as condescending, it is more accurate and less demonizing to say that someone is misguided in their belief than to betray them as heartless murderers. You could say, “Pro-choice people are people of deep moral conviction who are seriously wrong about the central issue of when life begins.” They could acknowledge that pro-choice people do not see themselves as murderers. You could acknowledge that a pro-choice person does indeed respect the sanctity of human life. Unfortunately, that doesn’t happen much if ever. I don’t want to hear prayer vigils that call for reversing Roe or for legislation outlawing abortion. I want to hear prayer vigils that people change their minds about when life begins.
Just once, I want to hear a reporter ask a pro-life person, “If you did not firmly believe that life begins at conception, would you find it cruel to force a woman to bear an unwanted child?” Similarly, I want reporters to ask pro-choice people, “If you were convinced that life began at conception, would you not agree that preserving that human is that life is more important than the personal choice of the woman who is carrying it?” We might find some common ground.
Ridicule of Pro-Choice
While the pro-life contingent demonizes pro-choice, pro-choice sadly often takes the stance of ridiculing the pro-life position. It is portrayed as absurd, ridiculous, dispassionate, and even ignorant. Some of it is rooted in the position that any religious belief is irrational and not to be taken seriously or respectfully. Liberals sometimes begrudgingly acknowledged that they respect people’s right to believe what they want to. They rightly point out that freedom of religion does not mean freedom to impose one’s religious views on others. However, much of the language and tone of the criticism leveled at pro-life is extremely condescending and couches the pro-life position as being irrational.
While it is true that the belief that human life begins at conception is a matter of faith, once you accept that a person’s belief is that life begins at conception, the pro-life argument is completely rational. I wish that the liberal position would acknowledge something along the lines of the following argument…
Suppose there was a law that nearly 70% of the population agreed to that said if, for some reason you didn’t want to raise your children, it was perfectly legal to murder them up until they were say one year old. One could argue that a one-year-old is incapable of sustaining their own life. In some ways without the support of another human being, it is not “viable.” The argument that “it’s my child and I can do with it whatever I want” would fall on deaf ears.
My guess is that liberals would not in any way shape or form support such a law. But they fail to knowledge that their moral outrage and revulsion at such a ridiculous idea is exactly the same as the moral outrage and revulsion that pro-life people have towards abortion.
Again, it’s a failure to recognize that both sides have the same sincere, deep-rooted respect for the sanctity of human life. If you really, really, really appreciated the idea that the debate is about when human life begins, then the pro-life position is just as morally upright as the pro-choice position. There is no basic difference in the morality of the two positions. Liberals fail to acknowledge that. Much of the derision that the left holds for the pro-life movement is a failure to understand or acknowledge that it really is (or ought to be) solely about when life begins.
If you truly believe that holding any religious belief is irrational, misguided, or ridiculous then have the courage to stand up and say so. Love him or hate him, comedian Bill Maher doesn’t hold back at all in his ridicule of any religious belief. I’m not saying that everyone who is pro-choice is anti-religion but many of their arguments against the pro-life movement are thinly veiled derision of religion and lack of respect for religious beliefs. I want to hear more acknowledgment from the left that the pro-life’s position is rational once one accepts the basic belief that life begins at conception. Go ahead and say, “You can’t impose your religious beliefs on me.” But don’t portray religious people as being ignorant, irrational, or dispassionate towards the plight of women unless they clearly are anti-women outside the abortion debate.
Hypocrisy of Pro-Choice
As I stated earlier, I’m not necessarily trying to change anyone’s opinion. I’m trying to be as fair and balanced as I can be. But I feel it necessary to point out what I believe is hypocrisy in the arguments from both sides. It’s going to be painfully obvious that I’m going to have more to say about the hypocrisy of pro-life than pro-choice. I’m sorry. That’s just the way I see it..
One of the things that disturbs me about the words “pro-choice” it seems that there are many women who choose abortion because they feel they have no other choice. On the liberal side of the debate, there seems to be a lot more energy expended on helping women choose abortion and not so much energy expended on helping women not choose abortion. In fairness, women’s right to choose not to have an abortion is not under attack. But I would feel much better about the liberal position they were more committed to promoting adoption. While the liberal stance is in support of things like childcare, equal pay for women, and other social justice issues that would make it easier for a woman to choose not to abort, there seems to be a fear on the part of the left that if they pay too much attention to helping women choose not to abort, that they will be branded as traitors to the cause for being too much “pro-life”.
In some ways, I’ve already covered some of the other hypocrisy of pro-choice. They tend to argue from a position of moral superiority and they ridicule the other side without acknowledging the morality and rationality behind much of the pro-life argument. And many, use their disdain for religion as part of the argument. Being mostly liberal myself, there may be other forms of hypocrisy that I am not sufficiently objective to realize. Sorry about that.
The Problem of Exceptions
One of the areas in which both sides show hypocrisy regards the issue of exceptions to abortion restrictions. The left tends to demonize pro-life for not allowing for exceptions. One of the problems is that pro-life is inconsistent in its views on exceptions. In many ways, the issue of exceptions is the biggest area of hypocrisy on the part of pro-life.
While the left demonizes the right for not allowing exceptions, in my opinion, if the right wanted to be logically consistent they would be even more inflexible about exceptions. If pro-life was really as pro-life as they claim to be, they would be even harsher on exceptions.
Let’s look at the exceptions one by one. Throughout this argument, we are going to take the position that human life begins at conception. After all, that’s my basic thesis that this is what the debate should be about.
The Life of the Mother
In my opinion if you really believe life begins at conception, then “the life of the mother” is the only, only, only acceptable exception in which abortion should be allowed. The rationale behind this is an argument for basic self-defense. Our common moral tradition is that one can defend one’s life with lethal force.
One of the things that muddies the water is that many times the phrasing of this exception is “the life or health of the mother.” That is lumping together two completely different things. If someone is going to make me sick, do I have the right to murder them in self-defense? I don’t think so.
The other problem with the “health” exception is that it is often undefinably broad. We need only look at the ease with which one can obtain medically necessary marijuana or a comfort companion pet to realize that it’s easy to get a doctor to sign off on lots of things based on one’s physical or mental well-being. That doesn’t mean I’m unsympathetic to the suffering that women go through during a problem pregnancy. It does not mean that I am unsympathetic to the emotional trauma that comes from having to endure nine months of pregnancy. But if one truly, truly, truly believes that human life begins at conception, is it morally justifiable to murder someone to protect your physical or mental health? If the health risks are extreme, severe, and reasonably likely to cause fatal harm then that’s different. And that goes for mental as well as physical health. If you are in such a mental state that the emotional trauma of a pregnancy credibly leads you to suicide then that is the life of the mother. Not the health of the mother.
Rape and Incest
First of all, let me acknowledge that I realize as a man I am uniquely incapable of understanding the trauma of these situations. No matter how sympathetic I try to be, it’s going to be insufficient. I know that. However, we are talking about what is or is not morally acceptable. Just because something is morally right doesn’t mean it’s easy. In fact, many issues of morality impose difficult and traumatic demands upon a person.
One of my biggest pet peeves about the entire abortion debate is lumping together the issues of rape and incest. Words have meaning. These are two entirely separate issues.
This is going to sound horrible but hear me out before you call me a monster.
Incest should never be an exception.
Let me explain. If adult close blood relatives conceive a child as a result of completely consensual sex, then there is no reason whatsoever that one should want to abort such a child.
”But wait a minute!” you will say. “Are you saying if an underage girl gets pregnant by her father, brother, uncle, or first cousin that she should not be able to abort the baby?” I did not say that at all. All of those scenarios are undeniably rape. They need to be acknowledged as rape even if they are consensual. “But”, you will argue, “Even if a girl is of legal age at 18 and consents to sex with a relative, it is likely that the power dynamic between the two in an incestuous relationship is wrong.” My response, “Yep… That’s rape and covered by the rape exception.”
I believe that lumping in incest with rape diminishes the fact that underage incest is rape and it fails to knowledge that as repugnant or taboo as we might feel a consensual incestuous relationship is, it is not rape. It is my opinion that consensual adult incest does not warrant an abortion. If one believes that human life begins at conception it certainly is not grounds to murder someone. And if someone believes as I do that abortion is simply a really bad idea it also is not grounds for abortion to be somehow a good idea.
“But wait a minute!” you will say. “What about the high probability of genetic abnormalities that comes from an incestuous conception?” I promise you I will address that in a later section.
Let’s get back to the topic of rape.
If we are going to take an absolute position that human life begins at conception, then you have to ask yourself, although rape is a horrible, unspeakable, unimaginable tragedy, does the fact that one is a product of rape make it justifiable to murder you. Forcing a woman to carry a child is a product of rape does indeed compound the pain and horror of the original experience and forces it to be lived continuously for nine months. But does the radically immoral act of rape justify the murder of an innocent human being? Most decidedly no.
It is a tragedy upon a tragedy upon a tragedy that a woman should have to suffer through such a pregnancy. But the murder of an innocent life compounds that tragedy even further.
If pro-life people were as pro-life as they claim to be. They would allow no such exception.
In a recent interview, Republican Gov Asa Hutchinson of Mississippi was asked about the fact that a restrictive abortion law he recently signed had no exceptions for rape or incest. In response, he reiterated his belief that human life begins at conception. Way to go governor. But then he said that he believed the issue of exceptions was still open to debate and strongly implied that he would support exceptions for rape and incest. Not out of sympathy for the victims of such crimes because of political expediency. He felt that it would be easier to get abortion restrictions passed if there were such exceptions. Given a choice between no restrictions on abortions and abortion restrictions with exceptions, it would reduce the number of abortions and to him that was acceptable.
My reaction to that was… “You fucking hypocrite!”
You can’t have it both ways. Either abortion is murder or it isn’t. Let’s go back to the example of the one-year-old. Suppose a rape victim carries the baby to term. The baby is born and after six months the mother decides they can’t stand it. Can they murder the baby because it prolongs the trauma of their rape? Is that justifiable homicide?
While I would have more respect for a radically pro-life person who had no exceptions except for the life of the mother (self-defense) and I would find their positions more logically self-consistent and not hypocritical if they would stick to their no exceptions policy, many of these idiots have had God awful ways of expressing that. Most notably are those who have said something along the lines of, “If a woman gets pregnant from a rape is God’s will.” Holy fucking shit! It’s no wonder that the left makes fun of the religious beliefs of such people.
Couldn’t you just say that the tragedy of rape should not be compounded by the tragedy of murder on top of that? Do you have to be so insanely tone deaf to imply that rape is God’s will? Not only do inarticulate and insensitive pro-life people damage their position and the movement through bad theology such as “it was God’s will she was raped“ they also engage in blatantly unscientific arguments such as “you can’t get pregnant from rape.” It takes a lot of ignorance to espouse bad theology and bad science at the same time but unfortunately, many politicians do.
If the left wants to be critical of the no exceptions policy, they need to say something like, “I understand that the pro-life people believe that abortion is murder and I understand that they believe that a child conceived of rape does not justify the murder of an innocent child. However, the pro-life movement seems insensitive to the plight of rape victims. I do not believe that life begins at conception and therefore that is why am in favor of abortion on demand and especially in the instances of rape.” But they don’t do that. Instead, they make fun of their religion. And in some respects, because the pro-life people are so inarticulate in expressing their position and so insensitive to the plight of rape victims that they hurt their own case irreparably. These radically insensitive inarticulate people besmirch the entire pro-life movement.
In all of the above I’ve been arguing from a radically inflexible position that life begins at conception and as I said at the beginning of the essay I do not hold that position. I explained my personal position that almost all abortion falls under that wimpy category of “a really bad idea.” In the instance of rape, given my total inability to understand or appreciate the trauma of a woman who has experienced such a tragedy believe I could give my wholehearted support to someone who wanted an abortion under such circumstances. If there is anything that turns abortion from a really bad idea into an acceptable idea, it’s the issue of rape. That’s my personal view.
Nazi Level Shit
In a science fiction story that I’ve not yet been able to get published, part of the story is of a young girl whose parents want to force her to have an abortion because of a unique genetic issue with the child. I won’t spoil the story by telling you what it was. In pleading her case before a judge, she says, “They are saying that people like me and my baby don’t have the right to exist. That’s some Nazi level shit!” While some people will argue that an old straight cisgender guy has no business trying to tell the story of a pregnant teenager with gender identity issues… the girl in the story is quoting me when she says that.
My disability is Spinal Muscular Atrophy Type 2. It is a genetic neuromuscular disease. It is possible to do amniocentesis and detect whether or not an unborn child will have SMA. I know for a fact that there are people who have done such genetic testing and based on the results have decided to terminate the pregnancy rather than give birth to a disabled child.
While I support a woman’s right to choose to terminate a pregnancy and while I have a general dislike for abortion and wish that women would not choose it, in the case of this particular type of abortion I cannot help but take a firmer stance. I take such things quite personally.
My disability is an integral part of me. While one might say, “What would my life be like if I had not had a spinal cord injury?” Or, “What if I had not suffered brain damage during a difficult childbirth resulting in cerebral palsy?” Or as was the case with many of my classmates who were slightly older than me, “What if I had not contracted polio resulting in my disability?” But one cannot say, “What would Chris Young be like if he didn’t have SMA?” In the case of a genetic disease, you cannot separate the person from the condition. You can treat the condition. Possibly come up with a medical cure. But Chris Young without SMA just can’t exist.
In general I don’t like abortion but I am much more likely to accept someone who aborts a child simply because they don’t want children, can’t afford them, it ruins their career or any number of reasons, then I could accept the idea of abortion based on genetic differences.
That’s some Nazi level shit.
I do however need to be fair in my portrayal of people who typically abort children with SMA. In the cases that I know of personally, these are couples who have already had a child born with SMA. They are typically SMA Type 1 which is considerably more severe than my Type 2. Up until recently, the life expectancy of a child with SMA Type 1 was under two years. Having already lost a child to the disease and faced with the possibility of having another child and losing them, they instead choose to abort. In other words, “let’s kill it now because it’s going to die eventually.” I can’t begin to imagine the tragedy of losing a child under any circumstances and I completely understand the desire not to suffer that tragedy again. However, I cannot condone such selective breeding. I find it not only personally insulting to me and people like me but I find it dehumanizing. It turns human beings into breeding stock.
The situation is even more complicated in that although prenatal testing can determine whether or not a child will have SMA, it cannot predict whether or not they will have the more severe Type 1 or a less severe Type 2 like I have or the even less severe Type 3. Furthermore, there are now treatments for SMA that if administered to newborns can in many cases allow them to grow up completely unaffected by the disease.
Despite my claims of Nazi-ism, I don’t condemn parents in that particular situation but I pray that they find other ways to enjoy the joys of parenthood without engaging in selective breeding. On the other hand, given the potential for abuse, there undoubtedly will be other people who choose to abort over other genetic issues that do not involve fatal disabilities. I cannot condone aborting a boy if you wanted a girl or vice versa. Do we abort blue-eyed kids or brown-haired kids or other trivial unwanted conditions? It’s a slippery slope.
That brings us back to the issue of consensual incest. Incestuous conception runs a high risk of genetic abnormalities as a result of doubling up recessive genetic traits. Whether we take the “human life begins at conception” objection to abortion or the “really bad idea” objection, I can’t condone aborting children based on the probability (or even the absolute certainty revealed by prenatal testing) of a genetic condition. Someone who engages in consensual incest and is unaware of the risks is behaving irresponsibly but that doesn’t justify abortion in my opinion. If human life does begin at conception, genetic abnormalities should not be grounds for murder.
A Rational, Non-Religious Argument That Human Life Begins at Conception
While we are discussing genetics, I want to put forth what I believe is a rational, non-religious, argument for the position that human life begins at conception. First, let’s look at a rather ridiculous and spurious argument against the idea.
Some pro-choice people take the view that it is ridiculous to afford human rights to a single fertilized ovum. They go on to speculate that why should we stop there? That every sperm and unfertilized ovum a potential human being to be regarded the same rights? The epitome of this stance was the satirical song by Monty Python “Every Sperm is Sacred.”
While many religious traditions are opposed to masturbation, I don’t think I’ve heard anyone who is pro-life who seriously wants to extend human rights to every sperm.
So what is the difference between unfertilized zygotes and a fertilized cell? The difference is the completeness of genetics. If I ever get around to writing my autobiography, I already know with the opening line is going to be.
Whether or not you believe I was a human being at the time, it was at the moment of my conception that it was determined that I would have the genetic neuromuscular disease known as Spinal Muscular Atrophy. That fact is influenced every aspect of my entire life in a unique way. It is an integral inseparable part of me.
It is at conception a unique combination of a single sperm with a single ova that defines us uniquely as the human being that we will come to be. Obviously, not everything that happens in our life is determined by genetics. But that part of us that makes us unique individuals begins at that moment.
I think it’s a pretty good argument. It doesn’t rely on “The church says so” or “My interpretation of the Bible says so.” It is probably more philosophy than science but there is a great deal of science involved in the argument. It is at the root of my opposition to abortion for purposes of selective breeding.
For me personally, it does not rise to the level of “abortion is murder.”
Is It a War on Women?
Part of me wants to be disturbed when the left characterizes the abortion debate as a “war on women.” The main problem is, that there a are number of men and women whom I love and respect who are very decidedly pro-life and are in no way misogynistic or anti-woman.
The prime example is my late mother. Sometime in the mid-1960s before Roe in 1973 and before it was commonplace to be politically active on the issue of abortion, my mom testified before the state legislature in support of an abortion ban. My family has been described as a perfect poster child for pro-life (although we’ve never been on a poster).That comment was made one Sunday when I, my mother, my grandmother, my sister Carol, and Carol’s daughter Britney presented the gifts at the offertory of a mass at St. Gabriel on Respect Life Sunday one time. I was born with a genetic disability and we’ve already established that some people consider that grounds for abortion. My mother gave birth to five other children after me all of which were born premature and none of which lived more than 48 hours. After that experience, we adopted my sister Carol through a Catholic adoption agency. Her biological mother could have chosen to abort her but did not. Carol’s daughter Britney (as well as 2 more daughters that would come later) were all born out of wedlock. Finally my grandmother Helen Osterman was approaching age 90 and had been chronically ill for the last five years of her life. I can’t speak for the rest of them, but my pro-life beliefs come not because the Catholic Church tells me so but because it resonates in my life and my family. Nothing about our pro-life stance is in any way disrespectful towards women.
I’m offended when pro-life is equated with misogyny.
As stated earlier, neither the sincere belief that human life begins at conception nor the less stringent belief that abortion is a bad intrinsically requires one to be opposed to women’s rights.
Sadly, it is painfully obvious that that is not the case in a large portion of the pro-life movement especially when it comes to male politicians. The fact that organizations like Planned Parenthood have only a small percentage of their activities related to abortion and the fact that federal money that goes to such organizations cannot and is not used for abortion services does not stop people from trying to destroy such organizations. The vast majority of the work of Planned Parenthood is dedicated to women’s health yet the pro-life advocates want to destroy the organization.
One somewhat valid argument goes something like this… If a neo-Nazi group or a KKK chapter was running a woman’s health clinic, even if they didn’t perform abortions would you be in favor of funding their health clinics considering their abhorrent positions in other areas? Of course not? So the fact that 95% of what Planned Parenthood does is not related to abortion, do we want to implicitly endorse an organization that murders children? Crazy as it seems, it’s not a bad argument.
Here’s the problem… If I was a rich white male politician with varied business interests who was legitimately pro-life and not anti-women, I would open up a string of women’s health clinics that provided all of the same non-abortion services provided by Planned Parenthood. I would suck away all of the government funding from Planned Parenthood into my little side business of taking care of women’s health because for me I don’t hate women. I just hate abortion. If that was the case… I would understand and appreciate the legitimacy of their opposition to Planned Parenthood.
But no one has done that. And for me, that is the real proof that a disturbingly large segment of the pro-life contingent is indeed inducting a war on women’s rights and women’s health. Part of the problem is that Planned Parenthood supports birth control. And although the Catholic Church is opposed to artificial birth control, most evangelical denominations on the religious right who are allegedly pro-life have no problem with birth control on moral grounds. However, that only applies to married women. To them, providing women’s health services is somehow condoning a promiscuous lifestyle. Even if it’s not about birth control, Planned Parenthood serves mostly poor people. And the conservatives cannot help themselves but look at poor women, especially single mothers (even if they chose not to have an abortion), and blame them for their condition. They are completely unsympathetic to the needs of such women.
Again, my proof that this is the case is the fact that no one has opened up a string of women’s health clinics that do everything except abortion.
I’m not painting the entire pro-life movement in these broad strokes. People like my mother and my family are not small exceptions to the rule of misogyny. There are lots of pro-life people who are supportive of women’s rights that do not have anything to do with abortion. But there are way too many male decision-makers and even some holier than now women who stereotype those who are in need of women’s health services as sluts. These same people are opposed to universal healthcare of any kind, oppose other social service programs such as food stamps, welfare, and disability programs, and are especially unsympathetic to immigration issues.
These are all Christian values that they ignore yet they are supported by religious fundamentalists simply because they claim to be pro-life. Such hypocrisy is a grave disservice the to the legitimate cause of a pro-life philosophy.
One of the Ten Commandments says, “You shall not take the name of the Lord in vain”. While many people interpret that to mean that you should not swear using God’s name, I’ve always understood it to mean that you should not call yourself Christian and then act otherwise. All you do is give legitimacy to those who say that all religious people are hypocrites. You damage the brand.
It is disturbing to me how little of the pro-life movement focuses its energy on helping women to keep their babies and to have the economic stability to raise them. In the same way that pro-choice focus not so much on helping women choose not to abort, the pro-life side expands a disproportionate amount of their energy on legislating against abortion rather than supporting the needs of women who choose not to abort. There is an attitude that once the baby is born, they have saved an innocent life and their work is done. I’m not saying everyone who is pro-life has that attitude but it is way too prevalent.
Pro-Life Self Delusion
I hate to beat up on one side more than the other. However, there is one more criticism and I have to level at a large piece of the pro-life movement.
I don’t think they are as pro-life as they claim to be. I’m not talking about the hypocrites who oppose women’s health services or any of the other disingenuous things that I described above. I’m talking about people who sincerely claim to be pro-life and sincerely claim that they firmly believe that human life begins at conception. But their actions are disproportionately insufficient to illustrate the sincerity of that belief.
Let’s go back to the example of the hypothetical law that would allow one to murder an unwanted child up until the age of 12 months. If such a law existed, what would be your response? Would you sit idly by and hold prayer vigils? Would you peacefully protest? Would you debate and advocate for your position or would you take action to save innocent lives?
I can in no way endorse violence. I don’t think blowing up abortion clinics is the answer or committing violence against those who participate in such facilities is appropriate. Then again, let’s take another hypothetical. Suppose it was a country that has a law permitting the murder of children up to a certain age. Would we go to war against that country? If not war, what about significant economic sanctions? Wouldn’t we do anything possible to prevent such an atrocity?
I think what I’m saying is that I have a deeper respect for those who picket up and down outside abortion clinics, chain themselves to the doors, and engage in other nonviolent acts of civil disobedience.
When I looked at my own beliefs against abortion, I realized that my opposition did not rise to the level of extreme acts of civil disobedience, constant protest, constant statements of radical outrage, and other extreme measures consistent with what I might do if one-year-olds were being slaughtered for no legitimate purpose simply because it’s what a woman chose to do.
So, I had a choice. Because I wasn’t sufficiently outraged that abortion on demand occurs in this country as a matter of law, it either meant that I was morally deficient for not caring or it meant that I really did not believe that a pre-viable fetus was a human being. After considerable self-reflection over the course of decades contrary to the teachings of the Catholic Church which I love and respect, I concluded it was the latter.
When I see people whom I love and respect who are decidedly pro-life and respectful of human rights and claim that they believe that human life begins at conception yet I do not see a reasonable expression of their outrage over abortion and I do not see action consistent with that level of outrage, I have to wonder do they really believe there is no difference between a pre-viable fetus and a human being which has been born? My guess is, subconsciously, they feel there is a difference. Deep down they do not believe that a fetus is a human being. If they did, they would be doing a lot more about it.
I’m not sitting in judgment of such people especially because I once was one.
I’m inviting them to look inside themselves as I did and see if they can find within themselves what I found within me. It is possible to be considerably, morally, opposed to abortion without the necessity to impose that belief on others because abortion is not really murder?
That’s a decision everyone has to make for themselves.
Is This The “United” States?
I’m not saying we all have to agree on everything. Disagreement and civilized debate is at the heart of American democracy. The reason I asked if we are “united” is the entire issue of states’ rights.
As I write this, a draft decision of the Supreme Court has been leaked and in all likelihood, Roe will be overturned. (I decided not to release this blog until the decision actually was really official.) The wording of that decision claims that it only applies to abortion yet the logic behind the decision is that there is nothing in the Constitution that specifically allows for abortion. Furthermore, the illegality of abortion has a long and ancient tradition. This implies that other rights such as interracial marriage, gay marriage, extramarital sex, and the use of birth control as well as other personal privacy rights which are not explicitly in the Constitution could be overturned as well.
Now that the draft document is finalized, it will be up to the individual states to decide whether or not abortion should be legal. The entire issue of states rights has always been a pet peeve of mine. I always believed I lived in the “United” States. I’ve especially experienced this problem when it comes to other social justice issues most notably healthcare and programs for people with disabilities. Depending on what state you live in, the kinds of programs and services that are available to you are inconsistent from state to state. Not only am I frustrated by states’ rights but by the entire issue of “local control” especially when it comes to school districts. At least in Indiana, and probably elsewhere, public schools are funded by property taxes. That means that poor inner-city school districts have less funding than more affluent suburban school districts. The quality of education that you receive depends upon your ZIP Code and the socioeconomic status of that ZIP Code.
One does not have to study history very deeply to understand that the issue of states’ rights and local control is deeply rooted in racism. It doesn’t take much of a leap to conclude that the variability of access to abortion once the decision goes back to the states will indeed have a racial component to it. And if not race, certainly socioeconomic status.
It saddens me to say this, but I would not be at all surprised if the United States of America is heading towards a second Civil War. The gullibility of American voters was amply demonstrated with the election of and the continued support for Donald Trump. The hypocrisy and lack of shame of mainstream Republican leadership will inevitably exploit the gullibility of the American public.
Russia didn’t interfere in our elections. The lack of respect for provable truth, common decency, the rule of law, and the willingness of unprincipled people to exploit it is what ruined our elections and threatens to do even more damage to democracy in the near future.
If such a Civil War happens, there will be those who blame it on abortion. Pro-life and pro-choice factions will continue to demonize one another and blame one another for the divisions in our country. And much of that will be because both sides failed to acknowledge what it is that they genuinely disagree about and they failed to acknowledge those on both sides of the debate who have a strong moral center and commitment to human rights.
It makes me sad.
One Last Disclaimer
One final time I want to reiterate that not everyone on either side of this debate is guilty of the accusations that I make. If you are offended by how I characterized either side, don’t presume I was talking about you unless you are guilty of my accusations.
Peace be with you.